Research
We are a research group focused on proteomics and mass spectrometry (Fig. 1). We aim to make progress in a number of areas:
1) Advanced proteomics via data independent acquisition (DIA) method development
2) Chemical proteomics and drug discovery
3) Targeted approaches to studying dynamics of protein-protein interactions
4) Global strategies for studying protein complexes based on cofractionation
5) Applications in host-pathogen interactions and innate immunity
Figure 1: Word cloud based on our paper titles and abstracts
1) Advanced proteomics via data independent acquisition (DIA) method development
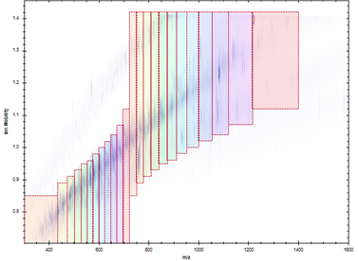
We have a long history of method development in DIA beginning in the early 2010s with Ruedi Aebersold’s group at ETH Zurich and the development of SWATH-MS. In particular we contribued to evaluting the inter-lab reproducility of SWATH-MS; computational strategies for generating spectral libraries; for analysis of DIA data; and for controlling error rates in DIA analysis. Most recently we co-developed the diaPASEF strategy with the labs of Florian Meier, Matthias Mann, Hannes Roest, Ruedi Aebersold, and Bruker Daltonics. This method integrates DIA acquisition with trapped ion mobility separations (TIMS) by synchronizing precusor selection with the release of ions from the TIMS cell as in the PASEF (parallel accumulation serial fragmentation) strategy, substantially improving performance over standard DIA methods. diaPASEF is our primary analysis strategy in current applied research and we aim to extend this method in future work.
2) Chemical proteomics and drug discovery
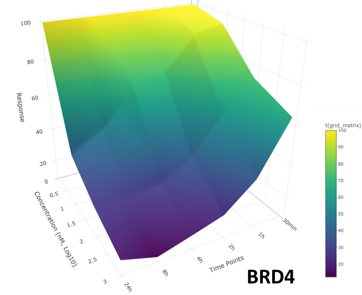
The targets of small molecule drugs are almost exclusively cellular proteins. However, most proteins expressed in human tissues are considered ‘undruggable’ due to the lack of ligandable pockets that are the targets of conventional small molecule drugs. New strategies to affect protein function in cells using small molecules are emerging that will have broad impact on basic biological research and drug discovery/development. These strategies include targeted degradation of proteins by induced proximity and covalent inhibitors. Technologies that the field of proteomics and chemical biology have developed in recent years are uniquely placed to support the development of drug discovery in these modalities. We have established a Chemoproteomics Centre of Excellence with the drug discovery company Almac Discovery along with partners across QUB. Our group is part of the Future Medicines Institute, A £55m investment in a new institute to help drive innovation and productivity in precision medicine in Northern Ireland, where we lead the proteomics platform.
3) Targeted approaches to studying dynamics of protein-protein interactions
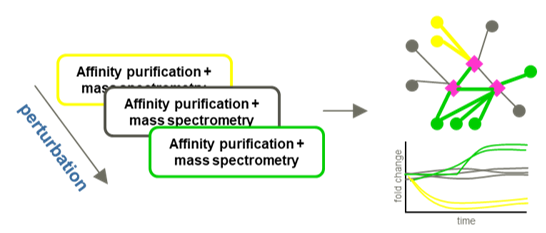
Protein complexes and protein-protein interaction (PPI) networks are essential mediators of most biological functions. Complexes supporting transient functions such as signal transduction processes are frequently subject to dynamic remodeling. Currently, the majority of studies into the composition of protein complexes are carried out by affinity purification and mass spectrometry (AP-MS) and present a static view of the system. To better understand inherently dynamic biological processes, methods to reliably quantify temporal changes of protein interaction networks are essential. We are using affinity purification combined with quantitative mass spectrometry to study dynamic reorganization of protein complexes. We have used this method study dynamic systems such as phosphorylation dependent PPIs in the 14-3-3 system and the connection between VCP/p97 mediated protein degradation and integrated stress response. Ongoing work will further develop these methods and apply them in complexes involved in host-pathogen interactions and innate immunity.
4) Global strategies for studying protein complexes based on cofractionation
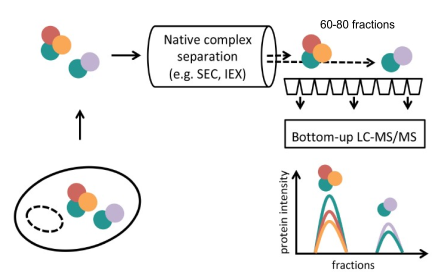
Targeted methods for studying PPIs such as affinity purification or proximity labeling are very powerful for focused study of particular systems. However, we also require unbiased methods that address the state and dynamic reorganization of protein complexes in cellular systems and can resolve PPIs to the level of protein complexes. This can be achieved using co-fractionation mass spectrometry (Cofrac-MS – a.k.a. protein correlation profiling – PCP, or SEC-SWATH). This method relies on the biochemical fractionation, frequently by size exclusion chromatography (SEC), of native cellular protein extracts combined with quantification of proteins by mass spectrometry (MS) analysis of each fraction. The concept rests on the idea that the composition of hundreds of protein complexes can be deduced by reconstructing and correlating the elution patterns of several thousand individual proteins across the protein complex fractionation space. Where two or more proteins co-elute, we take this as evidence of a protein-protein interaction or complex. This method has the attractive property that it can capture a global and quantitative snapshot of the proteome-wide organization of any biological sample from which native complexes can be extracted. We have participated in work to improve this strategy by combining with DIA acquistion and implementing a complex-centric data analysis workflow that uses prior knowledge about protein interactions as hypotheses to test in the CoFrac-MS data; establishing quantitative workflows that allow comparisons between conditions such as cell cycle; a network-centric approach to analysizing comparative data; and an approach to resolve differential partitioning of proteoforms into different protein complexes
5) Applications in host-pathogen interactions and innate immunity
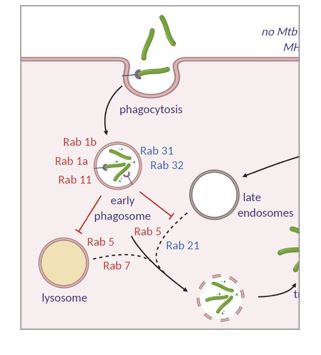
In recent years we have turned much of our attention to using the tools we have helped to develop in a more applied way. Specifically we are interested in the utility of these approaches in host-pathogen interactions and innate immunity. We have participated in projects characterizing the proteome of Mycobacterium tuberculosis in stress conditions as well as the effect of genetic variation on the quantitative proteomes in clinical isolates strains. Ongoing work is examining the interactome of bacterial effector proteins in host cells. We also aim to use our methods for characterizing reorganization of scaffolding complexes in the context of innate immune function.